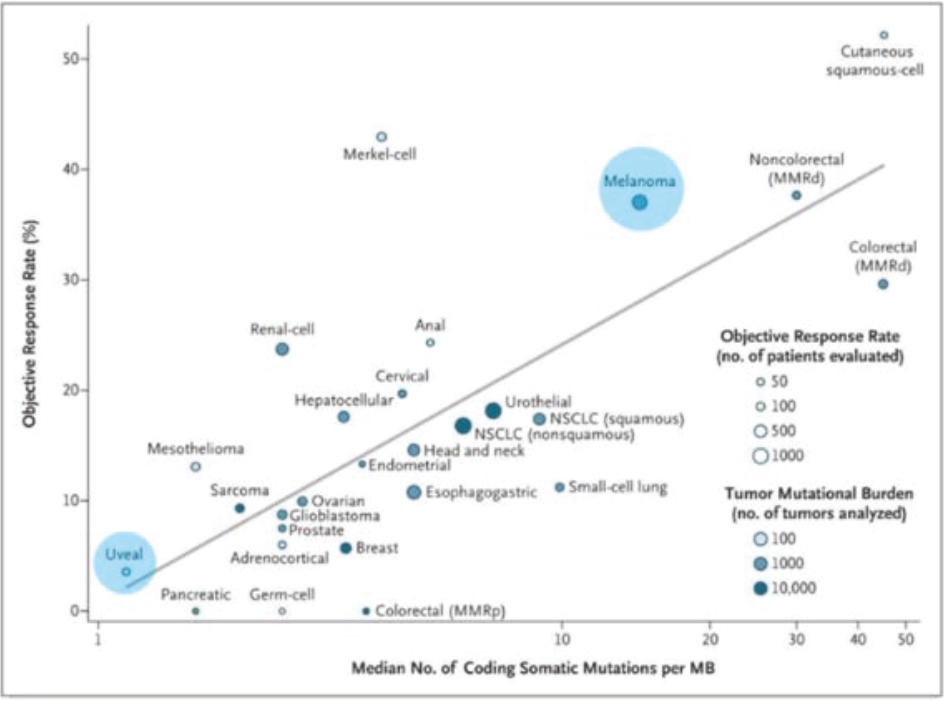
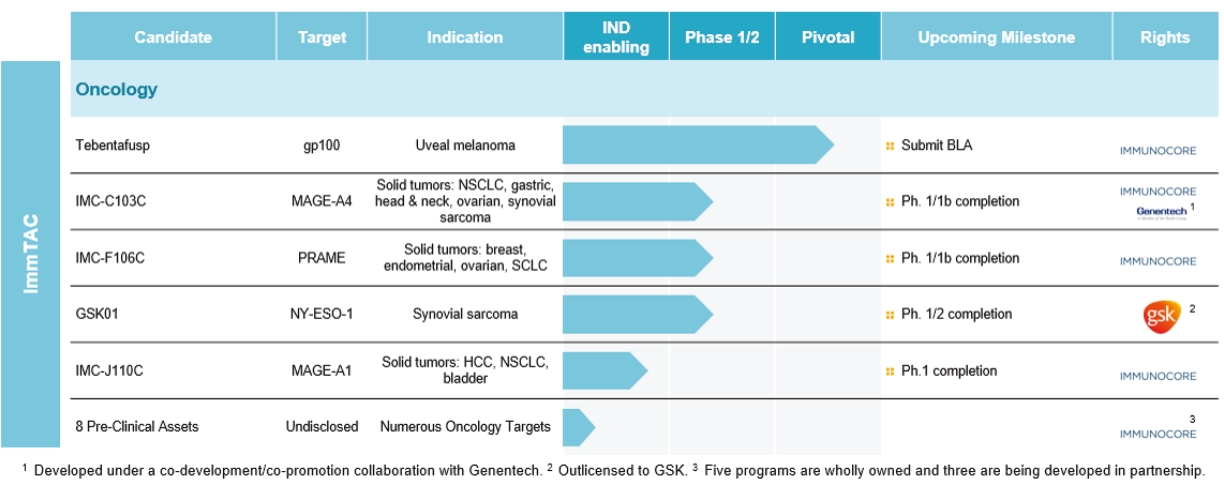
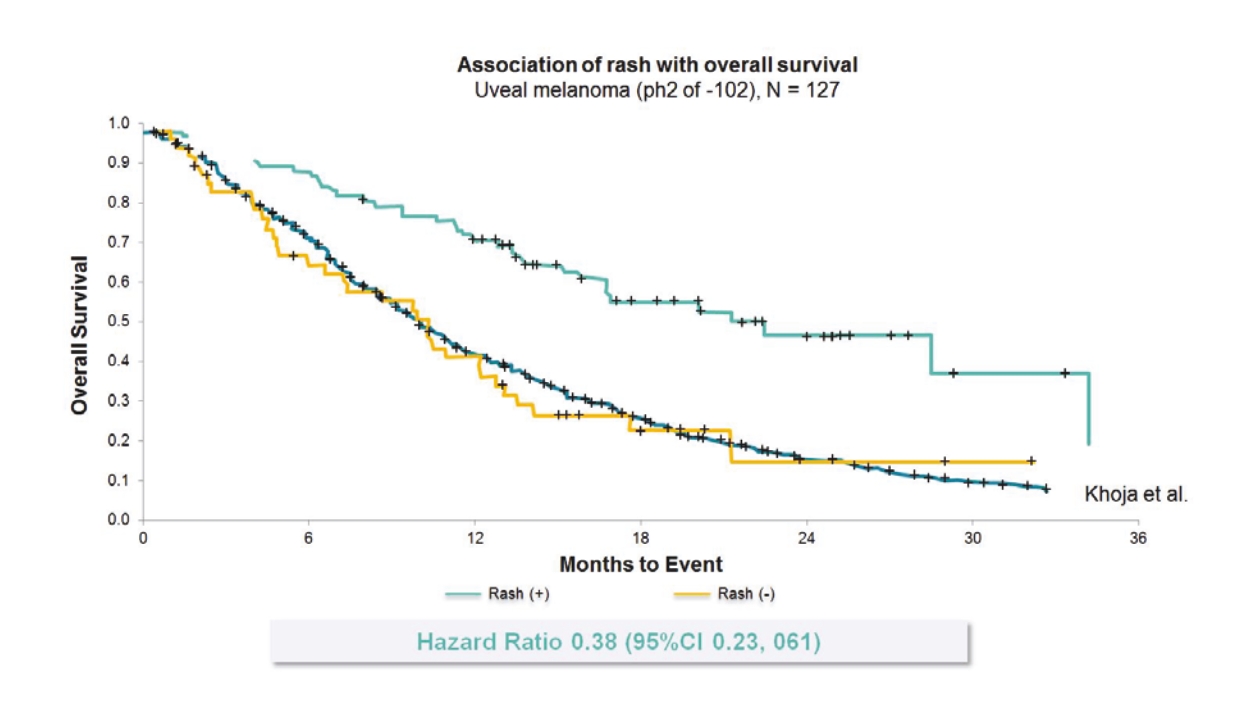
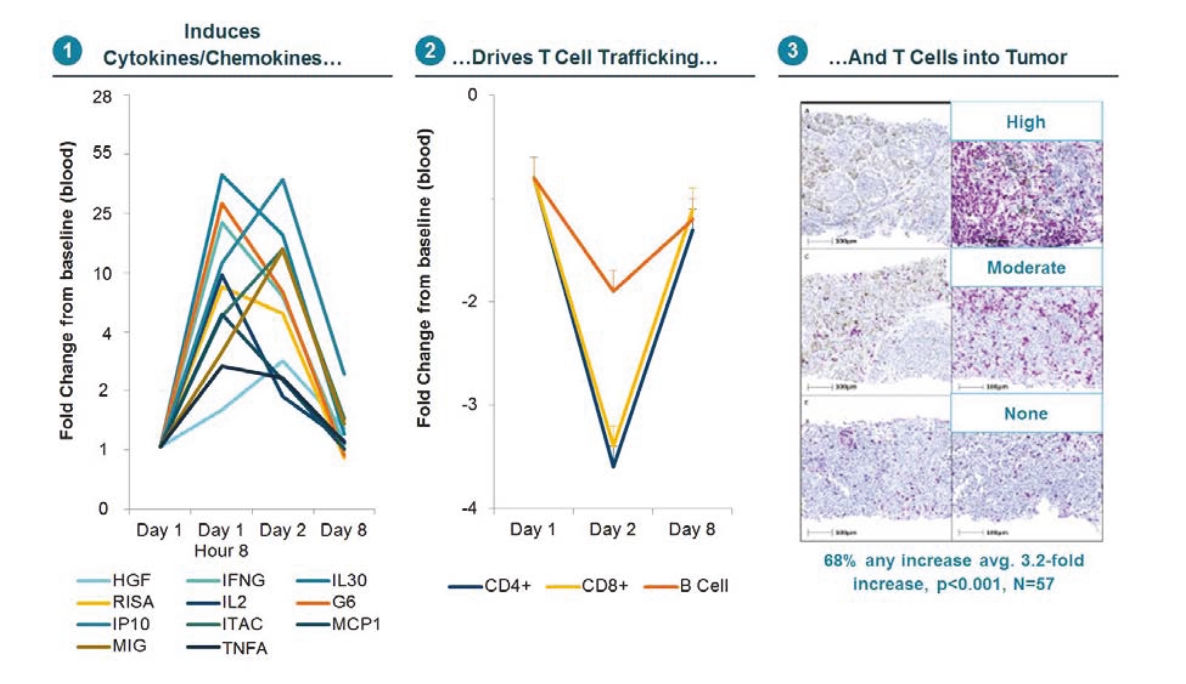
any of the physicians or other providers or entities with whom we expect to do business is found not to be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
We may not be able to file applications to commence additional clinical trials on the timelines we expect, and even if we are able to, the FDA or applicable competent authorities may not permit us to proceed.
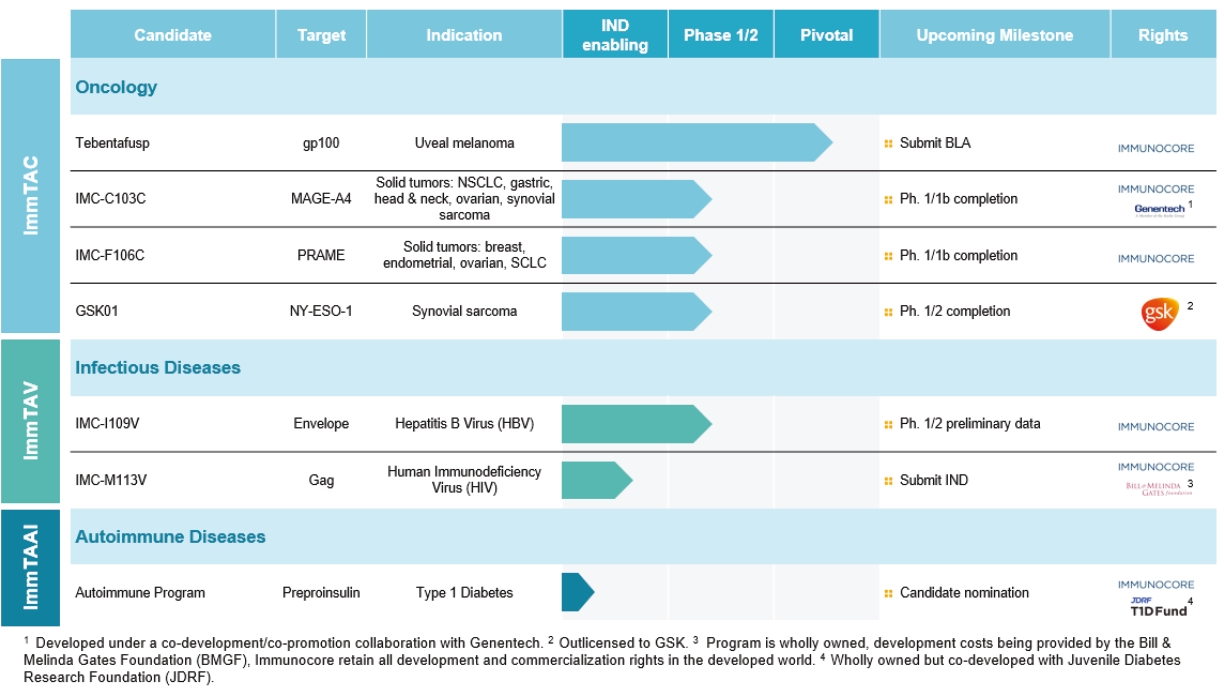
We plan to submit investigational new drug applications, or INDs, for additional product candidates to the FDA in the future. We also plan to submit applications to start clinical trials of additional product candidates outside the U.S. to the national competent authorities (for example, a clinical trial authorization, or CTA, to Medicines and Healthcare products Regulatory Agency, or MHRA, in the United Kingdom).
The filing of INDs to the FDA and the filing of applications outside the U.S. is dependent on additional data that have to be generated to support such regulatory filings. Hence, these filings may be delayed if the tests to generate those data show unexpected results or if technical issues arise in generating those data in the first place.
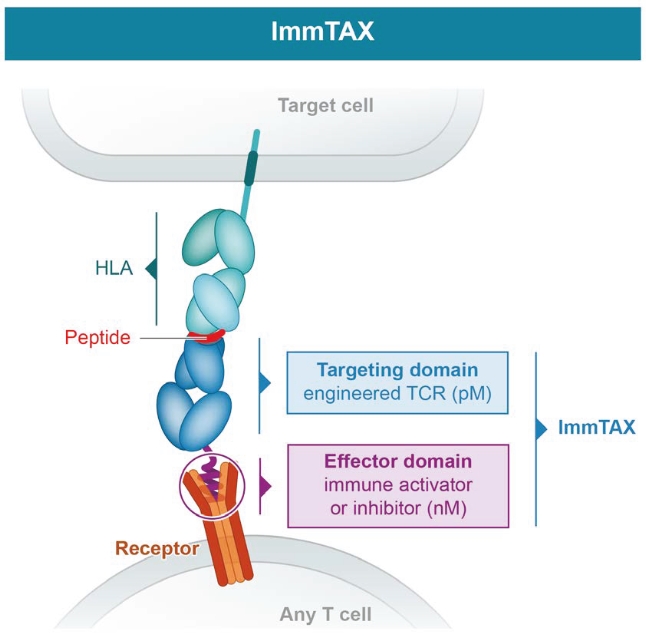
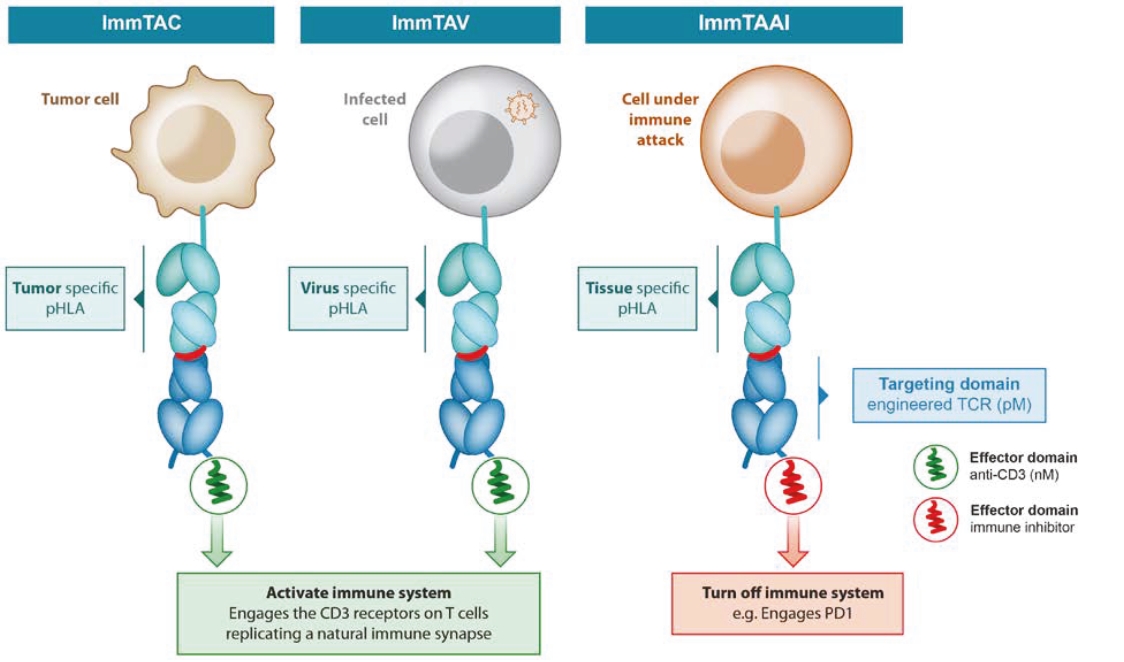
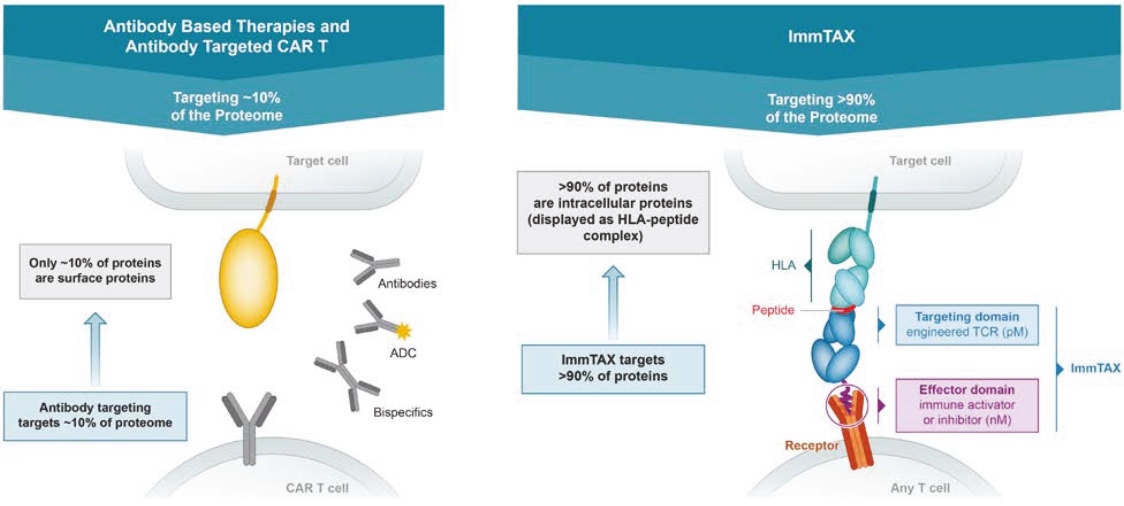
We cannot be sure that submission of an IND, IND amendment or CTA will result in the FDA or any other competent authority outside the U.S. allowing testing and clinical trials to begin, or that, once begun, issues will not arise that suspend or terminate such clinical trials. The manufacturing and pre-clinical safety and efficacy testing requirements of both ImmTAC® and ImmTAAI® remain emerging and evolving fields. Accordingly, we expect chemistry, manufacturing and control related topics, including product specification, as well as pre-clinical safety testing, will be a focus of IND reviews, which may delay the allowance of INDs by the FDA or CTA approval by other competent authorities outside the U.S.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including chemicals and biological and radioactive materials. Our operations also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological, hazardous or radioactive materials.
Changes in funding for the FDA, the SEC and other government agencies could hinder their ability to hire and retain key leadership and other personnel, prevent new products and services from being developed or commercialized in a timely manner or otherwise prevent those agencies from performing normal functions on which the operation of our business may rely, which could negatively impact our business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept payment of user fees, and statutory, regulatory, and policy changes. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of the FDA and other government agencies on which our operations may rely are subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new drugs to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, over the last several years, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA and the SEC, have had to furlough critical FDA and other government employees and stop critical activities. If a prolonged government shutdown occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business.